
生物信息学/单细胞转录组在线分析工具
IRIS3:单细胞转录组细胞类型分析
[编辑]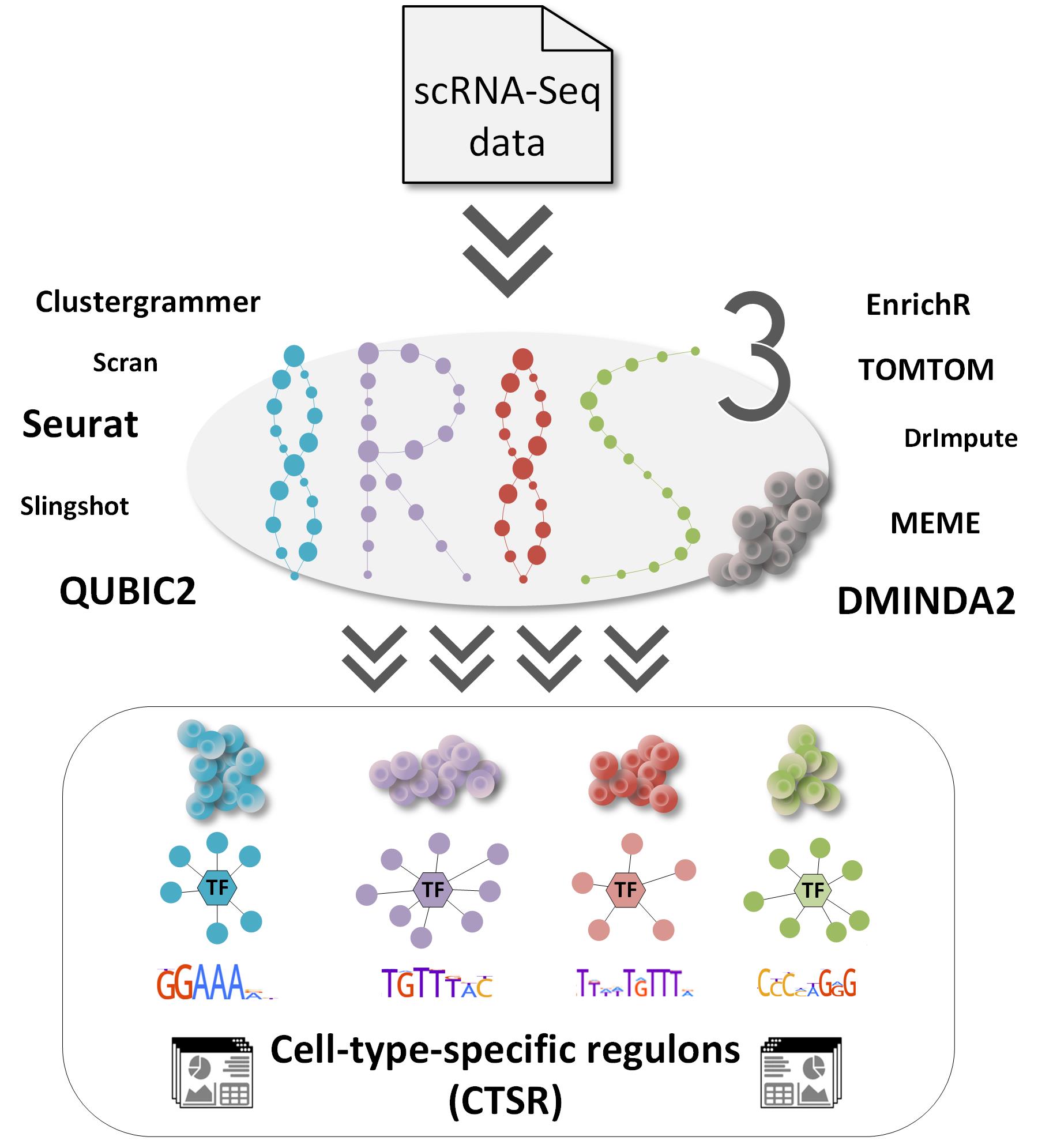{kind=link}
IRIS3(单细胞RNA-Seq的特定细胞类型调控因子推断服务器)是一个集成的Web服务器,根据人或小鼠单细胞RNA-Seq数据预测特定细胞类型的调控因子(CTSR,cell-type-specific regulon)。 CTSR能够可靠地识别和区分细胞类型,用于生物医学研究的计算或与实验分析相结合。 这些CTSR可以帮助阐明调节机制,并允许可靠地构建以特定细胞类型编码的全局转录调节网络。 IRIS3包括复杂疾病的层次结构和异质性,基因调节网络构建以及药物治疗的开发。
IRIS3在线工具:IRIS3。
IRIS3的功能
[编辑](1)它是一个提供CTSR识别的多合一框架,并结合了细胞类型特异性基因模块检测的双聚类分析和从头motif预测潜在的新型调节子;
(2)提供信息丰富的注释,支持对异质性调控机制进行深入分析;
(3)用户友好的Web界面,无需编程知识,具有简单的提交过程,全面的scRNA-Seq数据分析功能和高度交互的可视化效果。
(4)改善了调控机制的阐明,并允许可靠地构建以特定细胞类型编码的全局转录调控网络。
输入文件
[编辑]- scRNA-Seq基因表达矩阵(必需)和有三个配套的文件(由cell ranger生成),建议使用压缩文件减少上传时间。
- 单个txt,tsv或csv格式的基因表达矩阵,可接受gzip格式压缩文件。
- HDF5格式的特征barcode矩阵。
- 三个gzip格式的压缩文件记录了10X基因组输出的barcode,特征和基因表达信息。
标识符可以表示为Gene Symbol(例如HSPA9),Ensembl Gene ID(例如ENSG00000113013)或Transcript ID(例如ENSMUST00000074805)。分别使用org.HS.eg.db R包和org.Mn.eg.db包对人和小鼠的参考基因组的基因进行注释。
- 细胞标签文件(可选):一个两列的矩阵,第一列是与基因表达文件完全匹配的细胞名称,第二列是真实的细胞簇。聚类标识符可以是术语(例如2_cell_stage,4_cell_stage)或数字(例如1,2)。细胞标签文件将用于评估预测的细胞类型(否则省略评估)和规律推断(或使用预测的细胞类型)。
- 自定义基因模块文件(可选):一个文本文件,每一列应包含一个基因列表。对于基因模块(从文献中收集或通过其他工具生成),这些结果被视为“模块特异标签”,并且在最后一个细胞类型旁边具有单独的选项卡,以显示热图和结果。除了调控子发现功能外,用户还可以将感兴趣的调控子与上传的基因模块的调控子进行比较,寻找相似性。
输入文件示例:单细胞转录组在线分析工具IRIS3_示例数据。
{kind=link}
分析流程
[编辑]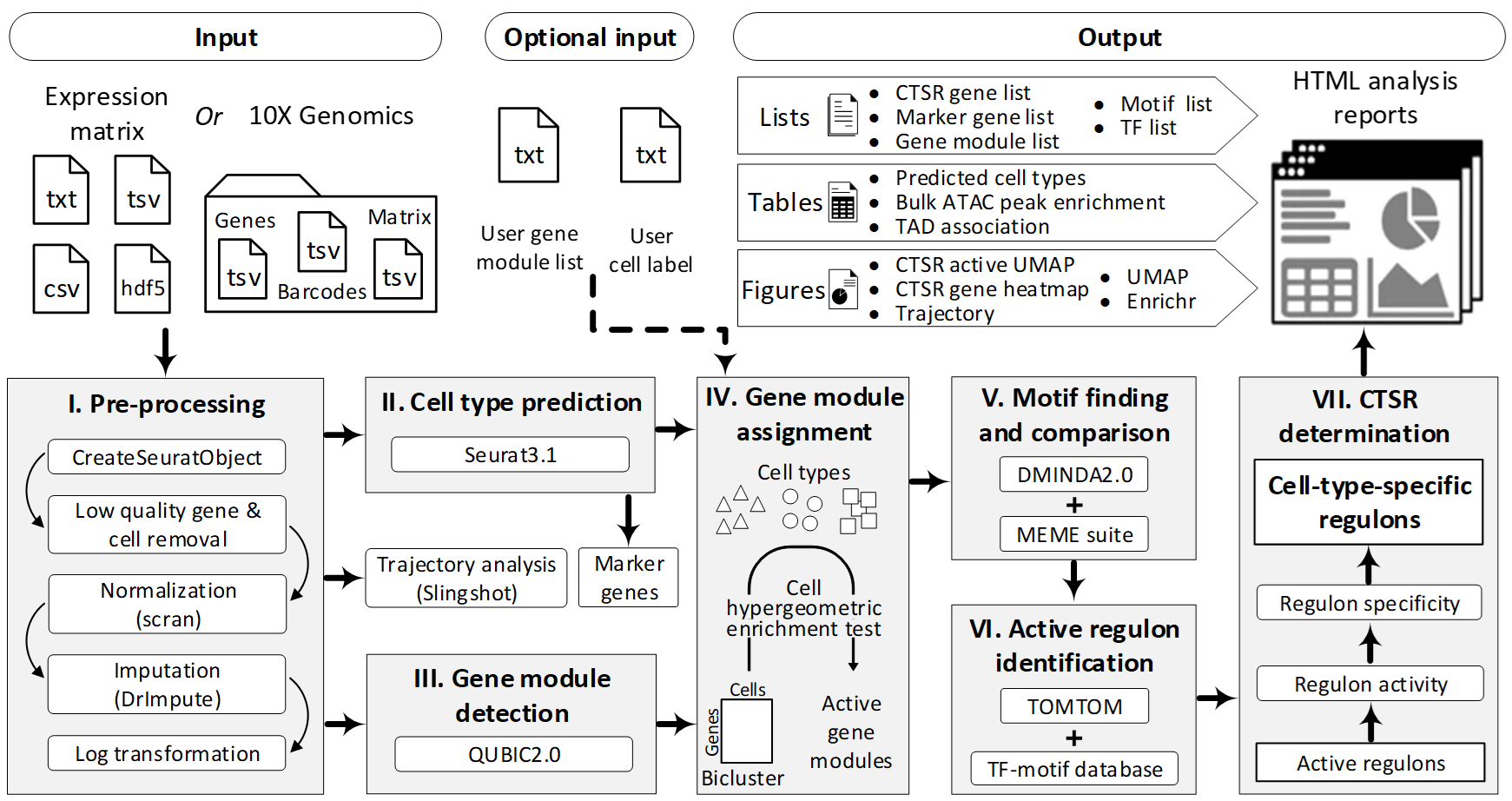{kind=link}
整个流程包括七个步骤:
IRIS3集成了多种最新工具,包括DrImpute,scran,Seurat,QUBIC2,DMINDA2和MEME。这些工具在分析领域中享有盛誉或已被证明具有最佳性能。
步骤一:数据预处理
[编辑]首先通过提交页面加载基因表达数据,然后创建Seurat对象。去除超过99.9%的细胞中具有零值的基因和具有少于200个非零表达基因的细胞,以提高分析性能。数据标准化状态是通过将整数视为未标准化的值自动检测的,而十进制已标准化。未标准化的数据将由scran标准化。缺失数据补全步骤可以根据用户的选择进行。最后,将表达式值进行对数log归一化log(x + 1)以重新缩放数据。
第二步:细胞聚类
[编辑]在Seurat(版本3.1)中可以预测细胞类型,并且大多数参数都设置为默认值。根据Seurat教程的建议,将主成分的数量设置为10,并为每个测试数据集绘制图形。通常,前十个主要成分可以覆盖数据变异的85%-95%,这些信息足够进行特征选择。然后,使用前十个主要成分进行聚类,聚类分辨率为0.8(Seurat中均为默认值)。值得注意的是,本研究以下各节中提到的细胞类型称为计算预测的细胞聚类。此步骤的输出是两列单细胞标签,将在步骤IV和其他轨迹分析中使用。
第三步:基因模块检测
[编辑]使用QUBIC2对来自步骤I的预处理数据进行分析,来检测基因模块。 QUBIC的先前版本已被证明是有效和高效地捕获高比例的二聚类(通过功能性生物通路富集的二聚类)中表现最好的方法之一。与QUBIC相比,QUIBIC2的性能有所提高。每个已识别的二聚体代表在特定细胞子集下的一组共表达的基因。
QUBIC2中解决了两个独特的功能:(1)它可以识别所有具有统计意义的双聚类,包括具有所谓“缩放模式”的双聚类,这个问题是相当具有挑战性的; (2)算法有效地解决了一般的双聚类问题,能够在几分钟内在数千种条件下解决成千上万个基因的双聚类问题。
三个主要参数控制IRIS3中的双聚类:
- 双聚类的重叠程度(从0到1)。 0表示没有重叠,而1表示完全重叠。默认值为0.7。
- 双簇识别的最大数量。默认值为500。较小的值可以充分减少运行时间,但可以标识较少的类别。
- 最小细胞数是双聚类块的最小列宽。预设值为20。
步骤四:确定活性基因模块
[编辑]如果双簇中的细胞与细胞类型簇中的细胞高度一致,认为双簇的组成基因会响应特定细胞类型中的调节信号。为了一致性,使用步骤II中识别出的二聚体的细胞成分(或上传的细胞类型)和步骤III中识别出的二聚体的细胞成分进行超几何富集测试。通过乘以N(cell type)x N(bicluster)将Bonferroni调整为与特定细胞类型相对应的bicluster的p值,其中N(cell type)表示细胞类型的数量,N(bicluster)表示bicluster的总数。如果超几何检验结果显著(adj.p <0.05),则认为双峰在相应的细胞类型中是活跃的。在双细胞群中的基因为该细胞类型的活性基因模块。
步骤五:主题查找和比较
[编辑]对于每种细胞类型,然后通过MEME和DMINDA2中的从头motif预测功能在活性基因模块中鉴定motif。使用hg38 / mm10参考基因组提取每个基因的上游启动子序列(默认为1,000 bp,可在提交页面上由用户设置)。人和小鼠的参考基因组分别整合在BSgenome.Hsapiens.UCSC.hg38和BSgenome.Mmusculus.UCSC.mm10 R程序包中。
第六步:调节Regruon
[编辑]使用TOMTOM将特定细胞类型中识别出的motif聚类,并用HOCOMOCO数据库(V11)中最匹配的已知motif进行注释。删除q值大于0.05的HOCOMOCO目标来过滤匹配的条目。 q值是最小的错误发现率,在该标准下,观察到的相似性是重要的。对于每个motif簇,相应的非冗余基因列表被命名为regulon。
第七步:CTSR推论
[编辑]对于每个调节子,细胞中的调节子活性得分(RAS)是基于所有相关基因在细胞中表达值的水平计算的。然后可以根据细胞类型内部细胞与外部细胞类型相比的RAS熵来计算细胞类型的regulon特异性评分(RSS)。 RSS的范围是0到1,值越高,表示此regulon在细胞类型中的特异性越高。通过将regulon的RSS与相同细胞类型中随机选择的基因集(通过bootstrap方法在regulon中具有相同数目的基因)的RSS进行比较10,000次,可以估算出regulon的RSS的经验p值。通过将细胞类型中所有已识别的调节子的数目乘以Bonferroni,可以调节Regulon p值。调整后的p值小于0.05的调节子是CTSR。有关调节调节子特异性得分的更多详细信息,请参见此处。
分析和可视化
[编辑]可以通过计算silhouette分数来评估预测的细胞类型(来自步骤II),该分数表示与其他聚类相比该细胞与其类型的相似程度。
如果用户提供了细胞类型,则预测标签还将通过调整后的兰德指数(ARI),兰德指数(RI),福克斯和马洛斯指数(FMI)和Jaccard指数(JI)进行评估。生成Sankey图来显示两个细胞组的收敛和发散(与Seurat预测对比)。箭头的宽度与flow成比例显示。有关细胞类型预测评估计算的更多详细信息。
针对不同的功能注释数据库执行富集分析,识别富集的GO功能,生物学通路等。富集测试由Enrichr执行。 Clustergrammer是由Ma'ayan Lab新开发的功能强大且用户友好的热图绘制工具,支持调控热图和相应的基因表达模式。
SC1:scRNA-Seq分析工具
[编辑]SC1是一个单细胞在线分析工具,包括质控,寻找Top基因,聚类,差异分析,基因富集分析,可视化(互动3D可视化,基因对,小提琴/柱状图,热图),细胞类型,TCR分析功能。
网址
[编辑]参考文献:M. Moussa and I.I. Mandoiu, SC1: A Tool for Interactive Web-Based Single Cell RNA-Seq Data Analysis, Proc. 16th International Symposium on Bioinformatics Research and Applications, pp. 389–397, 2020, pdf preprint, publisher url, bibtex
输入文件
[编辑]- 第一种格式:10x genomics的三个gzip格式的压缩文件(由cell ranger生成)。
- 第二种格式:单细胞基因表达矩阵,行为基因名,列为细胞类型。
示例数据
[编辑]SCANNER:单细胞转录组数据的注释,可视化,分享
[编辑]官网:SCANNER
输入数据的制作:Seurat流程
参考文献:SCANNER: A Web Resource for Annotation, Visualization and Sharing of Single Cell RNA-seq Data
alona:单细胞转录组在线工具
[编辑]网址:alona
alona是基于adobo包构建,该包基于Python语言构建了一套单细胞RNA-seq的分析框架。
细胞类型注释使用的标签基因来自PanglaoDB数据库。
参考文献:O. Franzén & J. Björkegren, alona: a web server for single cell RNA-seq analysis, Bioinformatics (2020), doi:10.1093/bioinformatics/btaa269
scQuery:综合单细胞转录组分析工具
[编辑]网址:scQuery
数据处理流程:sc-rna-seq-pipeline,将数据处理成HDF5格式的表达矩阵。
参考文献:A web server for comparative analysis of single-cell RNA-seq data